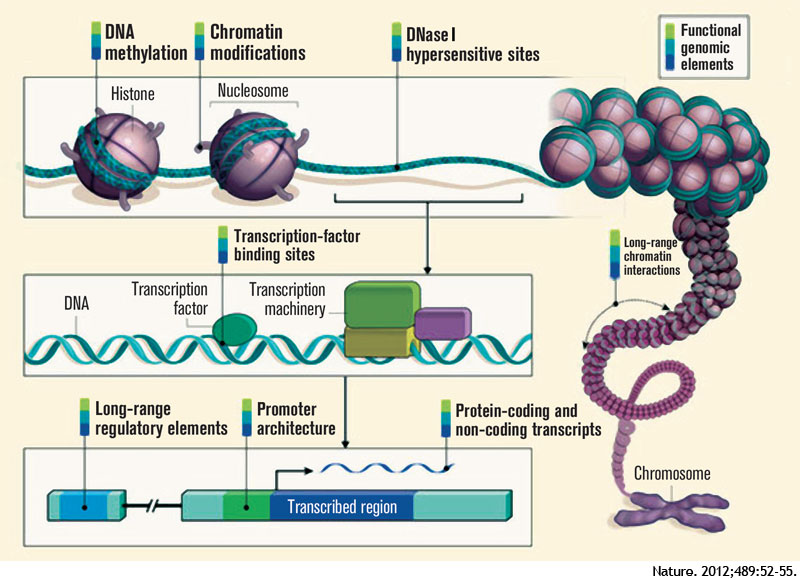
The ENCODE project provides information beyond that contained within the DNA sequence—it describes the functional genomic elements. The project contains data about the degree of DNA methylation and chemical modifications to histones that can influence the rate of transcription of DNA into RNA molecules (histones are the proteins around which DNA is wound to form chromatin). ENCODE also examines long-range chromatin interactions, such as looping, that alter the relative proximities of different chromosomal regions in three dimensions and also affect transcription. The project describes the binding activity of transcription-factor proteins and the location and sequence of gene-regulatory DNA elements, which include the promoter region upstream of the point at which transcription of an RNA molecule begins, and distant, long-range regulatory elements. One section of the project mapped DNase I hypersensitive sites, indicating specific sequences at which the binding of transcription factors and transcription-machinery proteins has caused nucleosome displacement.
Advances in genomics and bioinformatics have provided critical new insights into our understanding of the human genome—especially in identifying protein-coding genes—yet questions remain, including the functionality of the large stretches of noncoding DNA. Recently scientists in 32 labs around the world, as part of the Encyclopedia of DNA Elements (ENCODE) project, have reported in a staggering 30 papers in the journals Nature, Genome Biology, and Genome Research that the widely held belief that large segments of the human genome consists of “junk” DNA is incorrect. Findings from the ENCODE project reveal that 80% of the noncoding DNA does indeed have a biochemical function in at least one cell type. Contained within the noncoding roadmap of the genome are promoters, enhancers, and regions that encode RNA transcripts that have regulatory functions but are not translated into proteins (see Figure 1). The implications of these findings are protean in helping us to understand human variation and how changes in the genome result in disease.
Genome-wide association studies (GWAS) have indicated that a large proportion of the single-nucleotide polymorphisms (SNPs) correlated with disease phenotype are actually located in introns or within noncoding sequences. When the ENCODE consortium examined more than 4,500 SNP phenotype associations over a range of human diseases, they determined that 12% of these SNPs overlap regions that contain transcription factors, and 34% overlap DNase I hypersensitive sites, which are transcriptionally active DNA. For example, the SNP rs11742570, which has been shown to be strongly associated with Crohn’s disease, overlaps a GATA2 transcription-factor–binding signal. These findings tell us that many disease-associated changes are not actually in the genes themselves, but rather in the regions that regulate the genes. Clearly, interpretation of GWAS results must consider both the coding and noncoding regions of the genome.