What options do we have for assessing the postmarketing safety of medical treatments?
In nearly all countries, there is a long tradition of spontaneous reporting to the health authorities of unexpected, serious adverse events. However, this form of data collection is susceptible to reporting bias, reflecting the prescribing physicians’ level of uncertainty regarding the safety of new agents. Nevertheless, spontaneous reporting systems are valuable for generating safety signals, although these signals need verification through appropriate cohort studies, in which patients are followed from the onset of exposure to the outcome. This approach would require a known denominator, a defined observation period, an adequate control population, and complete ascertainment of adverse events.
Biologics registries that adhere to these standards have been established in several countries. Moreover, some northern European countries have the capability of linking their data to other national registries, which allows for complete coverage of outcomes (e.g., cancer or death).
Within the last year, three large European registries have published new data on the occurrence of solid malignancies during treatment with TNF inhibitors, describing the incidence and the recurrence rates of tumors. We think it is worthwhile to look at the common messages these data convey.
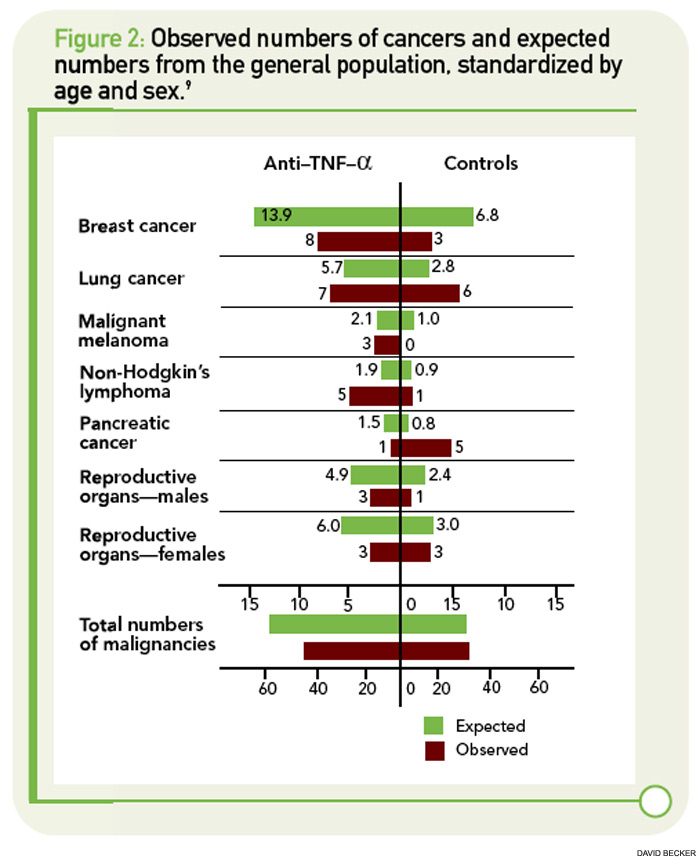
In Arthritis & Rheumatism, Askling et al reported on solid malignancies with data from the Swedish biologics registry ARTIS (See Figure 1, p. 34).8 The researchers linked data from ARTIS with population-based hospitalization and outpatient registries, as well as with the national cancer registry. A total of 67,743 rheumatoid arthritis (RA) patients were available as controls for the 6,366 RA patients treated with biologic agents. Because the reporting of incident malignancies in the cancer registry has been mandatory in Sweden since 1958, the investigators were able to identify each incident cancer in their biologics cohort. The total observation time in the ARTIS cohort was 25,693 person-years of follow-up (mean, 3.9 years).
The overall incidence of cancer was 9.3 per 1,000 patient-years of follow-up in the ARTIS cohort of patients treated with biologic agents. The relative risks of cancer were 1.14 (95% CI 1.00–1.30) compared with the general population, 0.99 (95% CI 0.79–1.24) compared with patients starting on methotrexate, and 0.97 (95% CI 0.69–1.36) compared with patients starting nonbiologic disease-modifying antirheumatic drug (DMARD) combination therapy. The incidence and relative risk of cancer did not change with observation time. Furthermore, the results were consistent, irrespective of the use of “time since initiation” of anti-TNF therapy or “cumulative duration of anti-TNF therapy” in the analysis.