Discussion
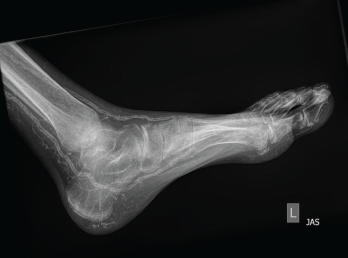
Figure 2: An X-ray of the left foot showed extensive vascular calcifications and no radiographic evidence of gout.
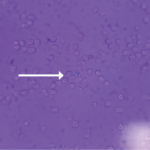
Crystalline arthropathies, such as gout, calcium pyrophosphate deposition (CPPD) disease and hydroxyapatite deposition, are not uncommon in patients with end-stage renal disease. The gold standard for crystalline diagnosis remains synovial fluid aspiration demonstrating the specific crystal, but in this case there was no joint effusion present for aspiration. The presence of monosodium urate (MSU) or CPPD crystals can be seen under polarized microscopy upon routine synovial fluid analysis, but other calcium crystals (e.g., hydroxyapatite and oxalate) are not typically reported in standard laboratory synovial fluid analysis and require additional stains (e.g., alizarin red) to distinguish crystalline etiology. Without confirmation of synovial fluid analysis, these crystal arthropathies can prove difficult to clinically distinguish from one another.
Gout is incredibly common in end-stage renal disease and is often easier to identify earlier in the course of the disease when it involves the first metatarsophalangeal joint. In renal failure, and as gout progresses, it can present as polyarticular and can be symmetric, involving the midfoot, ankle, knee and elbow.2