Treating Acute Episodes
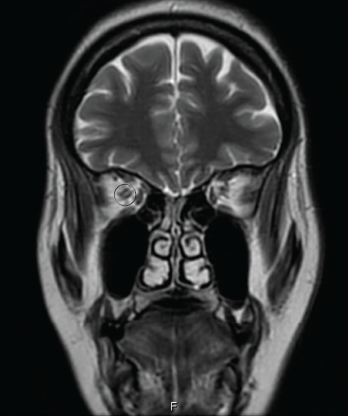
Figure 2: AN ORBITAL MRI
The goals of acute treatment are to suppress the acute inflammatory attack, minimize CNS damage and maintain long-term neurological function. Pulse-dose corticosteroids (e.g., 1,000 mg of IV methylprednisolone) daily for five days, followed by a two- to six-month taper, serve as initial therapy. Plasmapheresis has shown to be effective in patients who are refractory or show minimal response to steroids, making it a good alternative in a patient with coexisting infections.11