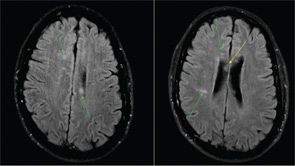
Figure 1: MRI of the brain with multiple foci of restricted diffusion (green arrows) primarily involving the white matter, notably, the central fibers of the corpus callosum (yellow).
The ophthalmology team was asked to evaluate SG, and a fluorescein angiography of the retina was performed (See Figure 2). A superotemporal branch retinal artery occlusion was seen with focal areas of nonperfusion in the right retina. There were associated areas of perivascular and retinal whitening with cotton wool spots. The occlusion was outside of the macula, which explains the preservation of vision in our patient. Initial imaging of the left retina also revealed an inflamed retinal arteriole; however, the examination could not be completed because of patient noncompliance.