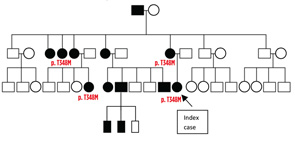
Figure 4: The family tree of an Israeli family with CAPS. Filled squares/circles indicate affected members. The affected members with genetic diagnosis are indicated with the mutation. Figure courtesy Dr. Yael Shinar, Sheba Medical Center, Tel-Hashomer, Israel.
When I (PJH) first saw the patient, it was clear she had hearing difficulties, and her mother had hearing aids in both ears. The patient also had an urticarial rash and red eyes. Genetic studies discovered the T348M pathogenic variant on one allele of the NLRP3 gene in the patient, her mother and other family members. Treatment with canakinumab led to a dramatic decrease in all symptoms except headaches, and she entered a rapid growth phase, including pubertal development. There was normalization of the acute-phase reactants. Her hearing loss did not progress.

