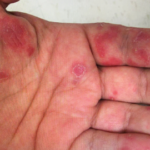
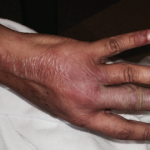
The case discussion also mentioned the patient had cutaneous changes suggestive of Rowell’s syndrome (i.e., erythema multiforme-like skin lesions occurring in context of anti-Ro/SS-A and/or anti-La/SS-B autoantibodies). Presumably that was a reference to the annular palmar skin lesions that were shown in one of the photos of this patient. However, on rare occasion, annular discoid lupus erythematosus skin lesions can preferentially localize to the palmar skin.
Another cause for refractory cutaneous lupus erythematosus is unrecognized drug-induced/drug-exacerbated cutaneous lupus erythematosus. SCLE is now known to commonly be triggered by delayed-in-time hypersensitivity reactions to members of a large number of prescription drug classes (e.g., thiazide diuretics, calcium channel blockers, ACE inhibitors, allylamine antifungals such as terbinafine, protein pump inhibitors, oncologic drugs and others). A list of drugs this patient was taking at the time of lupus erythematosus skin disease onset was not provided in the case summary. In addition to prescription drugs, one must consider certain over-the-counter drugs. As an example, physicians who start patients on prednisone often have the patient start taking an over-the-counter protein pump inhibitor, such as omeprazole, to minimize gastrointestinal side effects due to prednisone. Protein pump inhibitors are one of the more common drug classes that can induce SCLE. However, drug-inducted discoid lupus erythematosus is much less common, if existent at all. Some such published cases of druginduced discoid lupus erythematosus appear to, in fact, be drug-induced SCLE cases.
It is somewhat anomalous that a patient with such active SLE serologies, leukopenia, thrombocytopenia and persistent hypocomplementemia would have no evidence of internal organ SLE activity or damage. In the context of anti-double-stranded DNA autoantibodies and hypocomplementemia, SLE patients have traditionally been thought to be at increased risk for lupus nephritis. One setting in which this SLE patient’s clinical constellation might occur would be in the setting of a genetic complement deficiency state.
Homozygous genetic deficiency of C1q, the first component of the classical complement pathway, is one of the strongest monogenetic associations of SLE. And, C1q-deficient SLE patients frequently exhibit photosensitive forms of cutaneous lupus erythematosus. In addition, genetic deficiency of the C2 and C4 complement components has been associated with SCLE and possibly with discoid lupus erythematosus as well. One might postulate that genetic deficiency of an early classical pathway complement component could be a risk factor for treatment-refractory cutaneous lupus erythematosus. Familial discoid lupus erythematosus patients with heterozygous C2 deficiency have been reported to have a clinical phenotype similar to that of the patient described by Dr. Shapiro.2 However, the limited anecdotal published literature in this area does not fully support nor refute this hypothesis.