
The current therapies available for treatment to prevent fracture from osteoporosis target pathways of bone remodeling. We need to understand the mechanisms of bone resorption and bone formation, and the cells that control these mechanisms in order to have an impact on fracture prevention.
Osteoclast Development
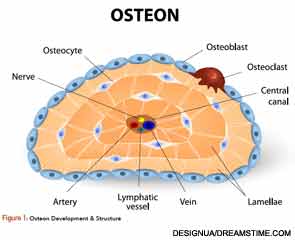
Osteoclasts (see Figure 1) are derived from hematopoietic stem cells, which when influenced by certain stimulatory factors, such as
macrophage-colony-stimulating factor (M-CSF), differentiate into monocytes. In the presence of receptor activator for nuclear factor kappa β ligand (RANKL) and M-CSF, they become multinucleated.
RANKL is essential for osteoclast formation. It is produced by osteoblasts and, ultimately, regulates bone remodeling.3 Osteoclasts attach to bone matrix, dissolving matrix and the organic part of bone, and producing tartrate-resistant acid phosphatase (TRAP), lysosomal enzymes, cathepsin K and integrins (see Figure 2). Osteoclasts express calcitonin receptors and RANK. This makes osteoclasts a major therapeutic target in osteoporosis.
To regulate osteoclast function, the formation, number, activity and lifespan of the osteoclast need to be controlled. This is accomplished through the RANK/RANKL/Osteoprotegerin (OPG) pathway. OPG is a cytokine receptor that is produced by osteoblasts and a member of the tumor necrosis factor (TNF) receptor family. OPG can decrease production of osteoclasts by inhibiting the differentiation of osteoclast precursors into osteoclasts and also regulate the resorption of osteoclasts in vitro and in vivo.
Osteoblasts, through RANKL and interleukin (IL) 1, IL-6 and TNF, activate osteoclasts to mature. When osteoclasts resorb, they release transforming growth factor beta (TGF-β) and insulin growth factors (IGF-1, IGF-2), which stimulate the osteoblast to mature and form bone.4
OPG production is stimulated in vivo by the female sex hormone estrogen, as well as the osteoporosis drug strontium ranelate.5 OPG has been used experimentally to decrease bone resorption in women with postmenopausal osteoporosis and in patients with lytic bone metastases. Another drug that can affect OPG is denosumab, because it can act as a decoy receptor for osteoblast-derived RANKL.