A cytotoxic or antiproliferative effect of methotrexate is clearly consistent with its known actions in the treatment of malignancies; however, methotrexate-mediated cytotoxicity cannot fully explain the antiinflammatory actions of methotrexate. Perhaps the most telling point in this regard is that, unlike the antiproliferative effects of methotrexate, neither folic acid nor folinic acid (except when given at the same time as methotrexate) consistently reduces or reverses the antiinflammatory effects of methotrexate in RA in randomized, blinded prospective trials.1 Moreover, the development of leukopenia, common in patients receiving high doses of methotrexate to treat malignancies, is a sign to cut back the dose or stop the use of methotrexate in treating patients with RA. Thus, other explanations for methotrexate’s mechanism of action have been explored (see Figure 1).
Figure 1
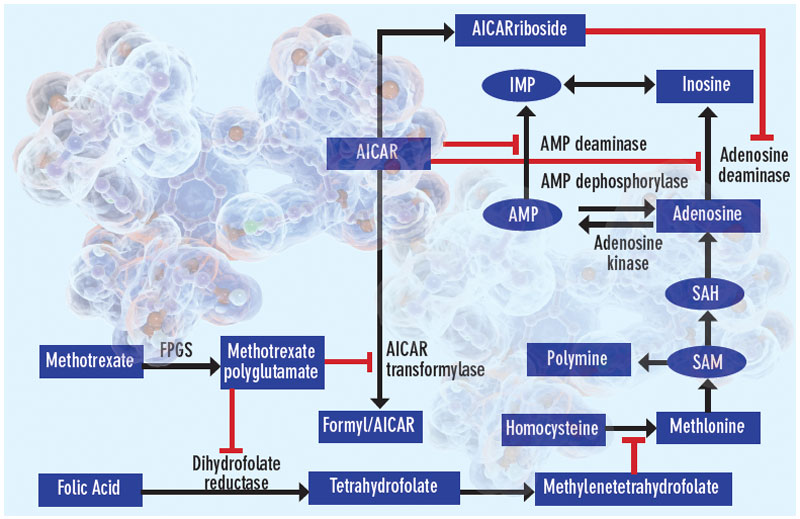
Proposed mechanisms of the antiinflammatory actions of methotrexate. Methotrexate exerts its antiinflammatory actions through a number of cellular mechanisms. Competitive inhibition of dihydrofolate reductase diminishes the de novo synthesis of purines and pyrimidines by preventing the regeneration from dihydrofolate of tetrahydrofolate, which is essential for the generation of folate cofactors required for purine and pyrimidine synthesis. Reduction of the levels of methyl donors, such as tetrahydrofolate and methyltetrahydrofolate, by the inhibition of dihydrofolate reductase results in the inhibition of the generation of lymphotoxic polyamines through methionine and SAM. Inhibition of AiCAr transformylase results in an increase in intracellular AiCAr levels. This increase has potent inhibitory effects on AMP deaminase and adenosine deaminase, which are involved in the catabolism of AMP and adenosine to IMP and inosine, respectively. The consequent accumulation of adenosine confers antiinflammatory effects. Levels of AiCArriboside, a metabolite of AiCAr, also accumulate and inhibit adenosine deaminase.
First Explanations
Among the first biological explanations put forth to explain the antiinflammatory action of methotrexate stems from the capacity of methotrexate to inhibit the regeneration of S-adenosyl methionine, the principal methyl donor in all cellular methylation reactions, from S-adenosyl homocysteine. Prior reports indicated that transmethylation reactions are required for the full inflammatory function of monocyte/macrophages, suggesting that inhibition of methylation reactions via methotrexate-mediated diminution of S-adenosyl methionine could explain the therapeutic effects of methotrexate.2 Moreover, inhibition of methylation reactions may result in the accumulation of intracellular polyamines, such as spermine and spermidine; these polyamines can be converted to lymphotoxic agents such as ammonia and hydrogen peroxide, thereby diminishing inflammation in RA. The hypothesis that methotrexate blocks inflammation by inhibiting the formation of methyltetrahydrofolate and causing the accumulation of polyamines was disproven in experiments using 3-deazo-adenosine. This agent is a specific methylation inhibitor but nevertheless fails to reduce inflammation in patients with RA despite inhibition of transmethylation reactions in their cells.3
Phillips and colleagues reported that methotrexate induces the formation of reactive oxygen species (ROS) in monocytoid and cytotoxic T-cell lines leading to their diminished function and death.4 These observations suggest another possible mechanism for the action of methotrexate. Since the effects on ROS required high concentrations of methotrexate and occurred primarily in rapidly dividing cells, their relevance to nondividing monocyte/macrophages is questionable. More recently, Spurlock and colleagues reported that methotrexate “primes” monocytes for apoptosis by a jun kinase–dependent mechanism that relies on methotrexate-dependent reactive oxygen species production and is reversed by tetrahydrobiopterin.5 Although the role of monocyte/macrophage apoptosis in suppressing inflammation in RA has not been demonstrated in vivo in either animals or patients, this work suggests an interesting mechanism by which methotrexate can act.
The doses of methotrexate used to treat RA are very low compared with those given for the treatment of cancer. Furthermore, it is clear from the once-weekly dosing schedule that the effects of the drug must be prolonged because serum methotrexate levels peak within six to 10 hours after an oral dose and are undetectable 24 hours after the dose. It has been known for many years that methotrexate is a “pro-drug”; methotrexate is taken up by cells and promptly polyglutamated by folylpolyglutamate synthase to long-lived methotrexate polyglutamates.6 The spectrum of enzymatic inhibition by methotrexate polyglutamates is different from the native agent, and the enzyme most potently inhibited by methotrexate polyglutamates is aminoimidazole carboxamidoribonucleotide (AICAR) transformylase, which catalyzes an intermediate step in de novo purine biosynthesis.
Inhibition of AICAR transformylase leads to intracellular accumulation of AICAR, a molecule with potent biologic effects such as inhibition of AMP deaminase, an enzyme which diminishes the intracellular concentration of AMP by converting it to IMP, and activation of AMP-activated kinase, a potent regulator of cellular metabolism.7 One consequence of AMP deaminase inhibition is the increased release of AMP into the extracellular space where it can be converted by the action of ecto-5’nucleotidase to adenosine, a labile but potent ligand for a family of receptors that regulate many different cellular and organ functions including inflammation.8
A Closer Look at Adenosine
My laboratory first reported that treatment with pharmacologically relevant doses of methotrexate promotes intracellular AICAR accumulation in mice and that AICAR accumulation is associated with elevated adenosine levels at inflamed sites.9 Moreover, my colleagues and I suggested that adenosine might mediate the antiinflammatory actions of methotrexate based on in vitro and in vivo evidence; subsequent animal studies have demonstrated that adenosine A2A and A3 receptors mediate the antiinflammatory effects of methotrexate.1,9,10 Further evidence that adenosine and its receptors mediate the antiinflammatory effects of methotrexate is provided by the observation that blockade of adenosine receptors by agents such as caffeine in animal models of arthritis and patients with RA reduces the antiinflammatory effects of methotrexate.11–13 The primary pharmacologic action of caffeine is blockade of adenosine receptors.
As noted above, adenosine is quite labile in biologic fluids (the half-life in blood is two to eight seconds) and is, therefore, difficult to measure directly.14 Therefore, to assess the role of adenosine in the response to methotrexate, Riksen and coworkers looked at an indirect measure of increased adenosine levels, evoked forearm vasodilation, and provided evidence that patients treated with methotrexate have increased adenosine responses.15 This finding provides further evidence for the hypothesis that adenosine mediates the antiinflammatory effects of methotrexate. Further support for this hypothesis has been provided by studies indicating that polymorphisms in genes involved in the pathway mediating adenosine production are associated with therapeutic responses to methotrexate.16-18
It can be truly said that the use of methotrexate to treat RA has transformed rheumatology as a specialty and raised therapeutic expectations for our patients and, for pharmaceutical companies developing new therapeutic agents, raised the bar for registration of new drugs.
Like all drugs, methotrexate has a range of toxicities. Clearly, as noted previously, the anemia, leukocytopenia, and hair loss that may accompany methotrexate therapy are likely due to methotrexate-mediated inhibition of cellular proliferation; these side effects can be prevented by appropriate use of folic or folinic acid supplementation. Other toxicities, such as severe fatigue on the day that the methotrexate is taken, rheumatoid nodulosis, and hepatic injury and fibrosis cannot be easily explained by the effects of methotrexate on cellular proliferation. Adenosine and adenosine receptor activation likely play a role in these toxicities; in the central nervous system (CNS), adenosine plays a central role in induction of sleep (hence, the capacity of caffeine, an adenosine receptor antagonist to prevent sleep) and it is likely that methotrexate-mediated adenosine release in the CNS leads to fatigue.19 Methotrexate promotes giant cell formation by cultured human peripheral blood monocytes, an in vitro model for rheumatoid nodule/granuloma formation, and this effect is mediated by adenosine A1 receptor activation.20 Finally, increased adenosine levels mediate hepatic fibrosis and steatosis via adenosine A1, A2A, and A2B receptors.21,22 Thus, methotrexate-mediated adenosine release likely plays a role in some of the toxicities of low dose methotrexate in patients with RA.
Methotrexate, one of the first drugs developed based on a specific chemical structure, turns out to have many different uses aside from that for which it was designed, treatment of cancer and related malignancies. A variety of pharmacologic actions explain the effects of methotrexate on RA and other inflammatory diseases, but the action for which there is the greatest amount of evidence is the promotion of adenosine release, which mediates the antiinflammatory effects of low-dose methotrexate via adenosine receptors.
Practice Implications for Methotrexate Use
The role of adenosine and its receptors in the actions of methotrexate suggests that ingestion of caffeine, an adenosine receptor antagonist, should be reduced or avoided in treating patients with RA. As noted previously, prospective studies demonstrate that caffeine ingestion diminishes the likelihood that methotrexate will be efficacious for the treatment of RA. In these studies, the effect of caffeine on the response to methotrexate was modest due, most likely, to caffeine’s relatively low affinity for adenosine receptors and its highly variable metabolism. Indeed, a large retrospective study of patients who had been on methotrexate for more than two years failed to show an effect of caffeine ingestion on the response to methotrexate.23 It should be noted that, in this study, many of the patients who experienced a poor response to methotrexate may have already been prescribed other therapies for their RA by the time the study was performed; this situation could have led to an enrichment of caffeine unresponsive patients in the cohort of patients studied.
The implication of these studies is that if patients with RA achieve a good response to methotrexate despite continued caffeine ingestion, there is no reason to stop their usage of coffee or caffeine-containing soft drinks; however, if the patients are not responding to methotrexate, it might be useful (if the patients tolerate it) to try abstaining from caffeine use for several weeks before adding another antirheumatic agent.
As has been well documented, the bioavailability of methotrexate diminishes at higher doses of the drug, a phenomenon most likely due to the fact that methotrexate is taken up from the gut by saturable transporters, which may be affected by luminal pH and other factors.24,25 In contrast, administration of either split dose or subcutaneous methotrexate leads to more reliable bioavailability of high doses of the drug; thus, for those patients who require higher doses of methotrexate to manage their RA, splitting oral doses of methotrexate or administering the drug subcutaneously might enhance patient responses to the drug.26-28
Acknowledgments
This work was supported by grants from the National Institutes of Health (AR56672, AR56672S1, and AR54897), the NYU-HHC Clinical and Translational Science Institute (UL1RR029893), and the Vilcek Foundation.
Dr. Cronstein is Paul R. Esserman Professor of Medicine; director, Clinical and Translational Science Institute; associate director for research, department of medicine; and director, translational medicine division, department of medicine, NYU School of Medicine, New York.
References
- Chan ES, Cronstein BN. Methotrexate—how does it really work? Nat Rev Rheumatol. 2010;6:175-178.
- Zimmerman TP, Iannone M, Wolberg G. 3-Deazaadenosine: S-adenosylhomocysteine hydrolase-independent mechanism of action in mouse lymphocytes. J Biol Chem. 1984;259:1122-1126.
- Smith DM, Johnson JA, Turner RA. Biochemical perturbations of BW 91Y (3-deazaadenosine) on human neutrophil chemotactic potential and lipid metabolism. Int J Tissue React. 1991;13:1-18.
- Phillips DC, Woollard KJ, Griffiths HR. The anti-inflammatory actions of methotrexate are critically dependent upon the production of reactive oxygen species. Br J Pharmacol. 2003;138:501-511.
- Spurlock CF, 3rd, Aune ZT, Tossberg JT, et al. Increased sensitivity to apoptosis induced by methotrexate is mediated by jun N-terminal kinase. Arthritis Rheum. 2011;63:2606-2616.
- Chabner BA, Allegra CJ, Curt GA, et al. Polyglutamation of methotrexate. Is methotrexate a prodrug? J Clin Invest. 1985;76:907-912.
- Corton JM, Gillespie JG, Hawley SA, Hardie DG. 5-aminoimidazole-4-carboxamide ribonucleoside. A specific method for activating AMP-activated protein kinase in intact cells? Eur J Biochem. 1995;229:558-565.
- Hasko G, Linden J, Cronstein B, Pacher P. Adenosine receptors: Therapeutic aspects for inflammatory and immune diseases. Nat Rev Drug Discov. 2008;7:759-770.
- Cronstein BN, Naime D, Ostad E. The antiinflammatory mechanism of methotrexate: Increased adenosine release at inflamed sites diminishes leukocyte accumulation in an in vivo model of inflammation. J Clin Invest. 1993;92:2675-2682.
- Cronstein BN, Eberle MA, Gruber HE, Levin RI. Methotrexate inhibits neutrophil function by stimulating adenosine release from connective tissue cells. Proc Natl Acad Sci USA. 1991;88:2441-2445.
- Silke C, Murphy MS, Buckley T, Busteed S, Molloy MG, Phelan M. The effects of caffeine ingestion on the efficacy of methotrexate. Rheumatology (Oxford). 2001;40:34.
- Nesher G, Mates M, Zevin S. Effect of caffeine consumption on efficacy of methotrexate in rheumatoid arthritis. Arthritis Rheum. 2003;48:571-572.
- Montesinos C, Yap JS, Desai A, Posadas I, McCrary CT, Cronstein BN. Reversal of the antiinflammatory effects of methotrexate by the nonselective adenosine receptor antagonists theophylline and caffeine. Evidence that the antiinflammatory effects of methotrexate are mediated via multiple adenosine receptors in rat adjuvant arthritis. Arthritis Rheum. 2000;43:656-663.
- Möser GH, Schrader J, Deussen A. Turnover of adenosine in plasma of human and dog blood. Am J Physiol. 1989;256:C799-C806.
- Riksen NP, Barrera P, van den Broek PH, van Riel PL, Smits P, Rongen GA. Methotrexate modulates the kinetics of adenosine in humans in vivo. Ann Rheum Dis. 2006;65:465-470.
- Sharma S, Das M, Kumar A, et al. Purine biosynthetic pathway genes and methotrexate response in rheumatoid arthritis patients among north Indians. Pharmacogenet Genomics. 2009;19:823-828.
- Wessels JA, van der Kooij SM, le Cessie S, et al. A clinical pharmacogenetic model to predict the efficacy of methotrexate monotherapy in recent-onset rheumatoid arthritis. Arthritis Rheum. 2007;56:1765-1775.
- Wessels JA, Kooloos WM, Jonge RD, et al. Relationship between genetic variants in the adenosine pathway and outcome of methotrexate treatment in patients with recent-onset rheumatoid arthritis. Arthritis Rheum. 2006;54:2830-2839.
- Diaz-Munoz M, Hernandez-Munoz R, Suarez J, et al. Correlation between blood adenosine metabolism and sleep in humans. Sleep Res Online. 1999;2:33-41.
- Merrill JT, Shen C, Schreibman D, et al. Adenosine A1 receptor promotion of multinucleated giant cell formation by human monocytes: A mechanism for methotrexate-induced nodulosis in rheumatoid arthritis. Arthritis Rheum. 1997;40:1308-1315.
- Chan ES, Montesinos MC, Fernandez P, et al. Adenosine A(2A) receptors play a role in the pathogenesis of hepatic cirrhosis. Br J Pharmacol. 2006;148:1144-1155.
- Peng Z, Borea PA, Wilder T, et al. Adenosine signaling contributes to ethanol-induced fatty liver in mice. J Clin Invest. 2009;119:582-594.
- Benito-Garcia E, Heller JE, Chibnik LB, et al. Dietary caffeine intake does not affect methotrexate efficacy in patients with rheumatoid arthritis. J Rheumatol. 2006;33:1275-1281.
- Hamilton RA, Kremer JM. Why intramuscular methotrexate may be more efficacious than oral dosing in patients with rheumatoid arthritis. Brit J Rheumatol. 1997;36:86-90.
- Yokooji T, Mori N, Murakami T. Site-specific contribution of proton-coupled folate transporter/haem carrier protein 1 in the intestinal absorption of methotrexate in rats. J Pharm Pharmacol. 2009;61:911-918.
- Tukova J, Chladek J, Nemcova D, Chladkova J, Dolezalova P. Methotrexate bioavailability after oral and subcutaneous dministration in children with juvenile idiopathic arthritis. Clin Exp Rheum. 2009;27:1047-1053.
- Hoekstra M, Haagsma C, Neef C, Proost J, Knuif A, van de Laar M. Splitting high-dose oral methotrexate improves bioavailability: A pharmacokinetic study in patients with rheumatoid arthritis. J Rheumatol. 2006;33:481-485.
- Hoekstra M, Haagsma C, Neef C, Proost J, Knuif A, van de Laar M. Bioavailability of higher dose methotrexate comparing oral and subcutaneous administration in patients with rheumatoid arthritis. J Rheumatol. 2004;31:645-648.