It starts with a Norwegian immunologist, Erik Thorsby, immunizing a colleague by transplanting skin and transferring cells from a healthy HLA identical donor. He assumed that this donor might possess a “hitherto-undetected HLA antigen.” In fact, his colleague, the recipient of the graft and known by his initials, FJH, produced an antibody that detected the new antigen, which Thorsby named, TH-FJH.
Apparently, Thorsby was not shy about using friends and families in his experiments. He subsequently tested his own family and noticed that one member who carried this new antigen had AS, while another possibly had this diagnosis as well.6 However, because most of his family members who were positive for this new antigen were completely healthy, Thorsby discarded this finding, assuming that it was caused by chance. I suspect that Thorsby’s family and friends were grateful that he reached this erroneous conclusion and abandoned his research before he could wreak havoc with their health.
A few years later, Derek Brewerton, MD, and colleagues at the Westminster Hospital in London, England, proposed to tissue-type patients with AS. Dr. Brewerton was intrigued by the clinical observation that AS could have a genetic predisposition. He was deeply influenced by the work of another colleague, Philip Gofton, MD, who described a strikingly high incidence of AS among the Haida Indians of the Queen Charlotte Islands in British Columbia, Canada.7
Dr. Brewerton reflected how this monumental study got started. “It was lunch in the common room at Westminster Hospital on a hot summer day in 1971. Over the salad we decided to investigate the frequency of HLA antigens in ankylosing spondylitis.”8 Although his proposal was initially turned down by his cash-strapped hospital, he eventually tested eight patients with AS for the HLA-B27 gene and found it present in all of them, a chance of less than one in a million. Bingo!
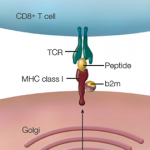
Following the discovery of this critical genetic association, which still represents the strongest link with any complex disease, there was a flurry of research attempting to explain the vital role of the B-27 antigen in the development of AS. Some investigators believed the gene provided some form of molecular mimicry, thus generating an aberrant the immune response. Others speculated a role for the gene in presenting arthritogenic peptides that could have been derived from enteric bacteria. More recently, the focus has turned to the strong genetic association between endoplasmic reticulum aminopeptidase 1 (ERAP1) with HLA-B27-positive AS.