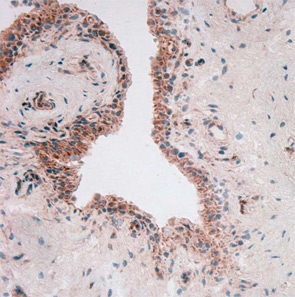
To throw adipose tissue–adipokine restriction dogma further overboard, another adipokine had to be introduced into this challenging scientific game: visfatin/PBEF. Lymphocyte aggregates and vascular endothelial cells predominantly synthesize visfatin in rheumatoid synovium (see Figure 2). Visfatin activates human leukocytes, induces co-stimulatory molecules on the cell surface of monocytes, and induces proinflammatory cytokines such as IL-1, TNF, and IL-6 in these cells; it also protects fibroblasts and neutrophils from apoptosis.7
Protective Adipokines
One question is frequently asked: Where are the “protective” adipokines? Similar to the proinflammatory cytokines, there should be at least one. So let’s therefore revisit the effects of “protective” leptin. The basis for this positioning is the observation that leptin-deficient ob/ob mice developed a less severe arthritis in comparison with wild-type mice. In humans, levels of leptin are positively correlated with the overall body fat content, but a true correlation between inflammation and leptin plasma concentrations is still under discussion.